
DNA解析が革新する生物学
今回は、神戸でNGS(次世代DNAシーケンサー)を中心に、コアファシリティとして神戸理研の研究を支える西村さんを、同じくコアファシリティとして研究基盤を支えている阿部さんから紹介されました。NGSは、多くの分子生物学で欠くことのできないツールになってきているけれど、BDRのコアファシリティではどんなことをしているんだろう。(聞き手:薬師寺秀樹)
もともとはタンパク質やってました
大学院の時は、伝統文化に関わる植物の研究をやってたんです。
へぇ。どんな?
ジーンズって超有名ですよね。でも、染料の藍色はむしろ日本の伝統文化なんです。その藍の研究をしていました。
確かに、比較的最近、大河ドラマで見た記憶があります。
タデアイという植物の葉っぱは見た目は緑色なんですけど、これをすりつぶすとインジゴブルーという真っ青な染料ができるんです。これが藍です。
どうやって青くなるんですかね。
葉の中にインジゴの材料になる無色透明な基質とその分解酵素があって、葉をすりつぶすと両者が出会って分解・酸化してインジゴブルーになります。いろいろ調べた結果、細胞の中の液胞という小器官に基質が入っていることが推測されました。それで、分解酵素をエレクトロポレーションで液胞に入れてやると見事に青い染料が出来て、細胞内の局在を証明できました。
エレクトロポレーションってあれですか、細胞に電気をかけてDNAとかを細胞内に取り込ませるやつ。
それです。タンパクを取り込ませる前例はありませんでしたし、細胞壁、細胞膜、液胞膜と三重の障壁があるのですが、試行錯誤してなんとかうまくいきました。その後、無酸素状態で実験が出来る装置などを自作して、今度は元になる基質と、その合成酵素を見つける事にも成功しました。
じゃあ、どちらかというとタンパク質の研究だったんですね。
そうなんです。でも、ちょうどその頃もうすぐヒトゲノムが公開されるらしい、そこではコンピューターが使われているらしい、という話を聞いて興味を持ちました。
2000年くらいですね。
今だと、生物学でコンピューターを使うことってごく普通だけど、当時、コンピューターはせいぜい論文なんかをワープロで書くのにしか使っていなかったんです。なにせ実験データはすべてアナログでしたし。
そうですねぇ。当時、図を描くのに苦戦して、最終的には手書きで紙を切り貼りしたりしてましたね。懐かしい。
そこにヒトゲノムという30億塩基対のDNA配列をコンピューターで解析するとか言われても、全然想像できない。当時は、たった一つの遺伝子のmRNAの配列をひいひい言いながら3、4カ月かけて決めていた頃ですからね。
別世界ですよね。
それでしばらく企業でIT関係の仕事をして、この世界に戻ってきました。
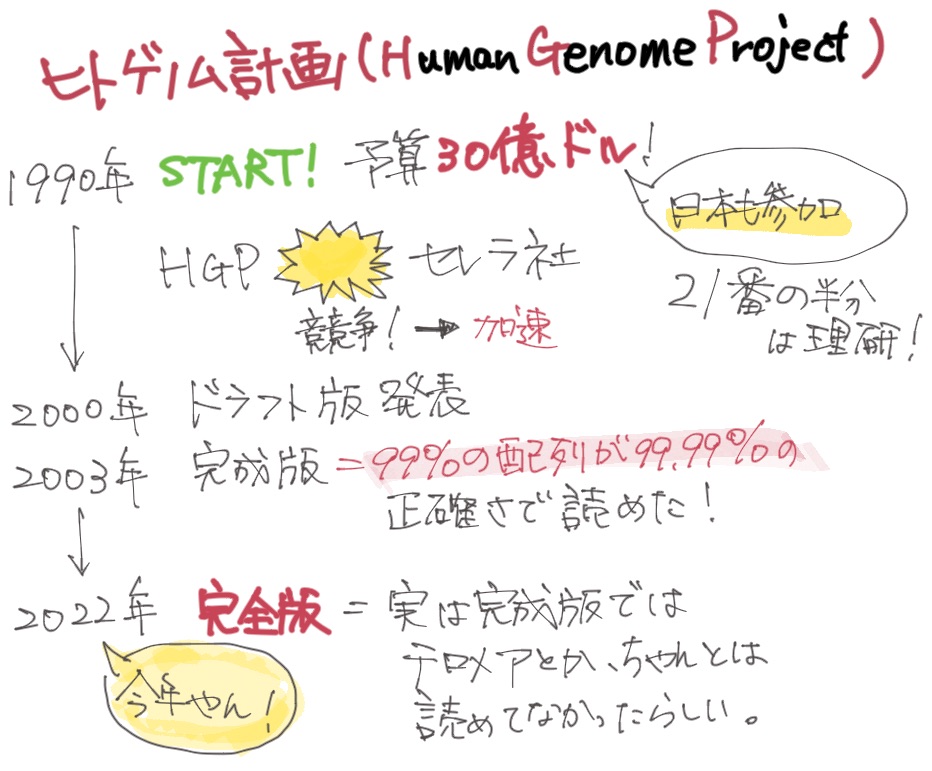
実はヒトゲノムがコンプリートしたのは今年!
その後は、DNAシーケンサーとゲノムというのは自分の研究のずっと芯にあります。
なるほど。
技術はどんどん進んでいくんですけど、NGSのすごさって、意外に世の中に伝わっていないと思うんです。
そうですか?
たぶん、僕のこの感動までは世の中に伝わっていないと思っています。
「感動」?
ヒトゲノムの解読っていつ終わったか知ってます?
2000年くらいですかね。ドラフトできたよーってクリントン大統領が発表したやつですよね。
ヒトゲノム計画というのは90年代に本格的に稼働するんですけど、それ以前にフレデリック・サンガーによってDNAシーケンス法が開発されて、いよいよヒトのゲノムが読めるかもしれないというのが20世紀の後半。
たしか、セレラという会社がシーケンサーをたくさん使って、国際プロジェクトに喧嘩を売った…。
で、急いで両者同時に「完了(仮)」ということでドラフト公開した、という感じですね。その後、完成版、完全版と続きます。
え?完全版?
今年の3月に公開されました。
最近やん!ドラフトから20年も経ってるじゃないですか!
ヒトゲノムって、読みにくい場所がたくさんあるんです。
というと?
ヒトゲノムDNAって、細胞のひとつひとつの核の中にあって、全長だと2mもあります。ものすごく長いわけです。染色体単位には分かれていますが、それでも長いので、端から端まで一気に読めない。だから、ゲノムDNAをぶつ切りにしてその断片を読んで、後でコンピューター上で繋ぐわけです。そのときに似たような配列があると、それが元々のゲノムのどこに位置するかがわからない。また、そもそも解読出来ない難読領域もあります。当時は技術的にそれ以上解決できないので、ひとまずその状態で公開されてたわけです。
あー、そうなんですね。知らなかった。
そういう読みにくい部分も全部きちんと読んで、完成したのが今年。”The complete sequence of a human genome”という論文タイトルでした。
コンプリート!
DNA解析の技術革新は止まらない
サンガー法は一つのDNAサンプルから色々な長さのDNAを合成させて、その末端に蛍光物質をつけて、長さ順に並ぶ電気泳動という方法を使って、一つずつ読んでいくわけです。
たくさんやると、ズレて間違えそう…。
そうなんです。しかも、当時は巨大なゲルを手作りして、泳動写真を人が目で見てましたし。
あ、そうか。デジカメなんかまだ使い物にならなかった頃か。
そうそう。で、その後キャピラリーという細い管の中で同じことができるようになって、配列解析が進んでいくことになります。キャピラリーは並列化がしやすいので、速度が大幅に上がりました。でも、ただ並列化しただけ、とも言える。
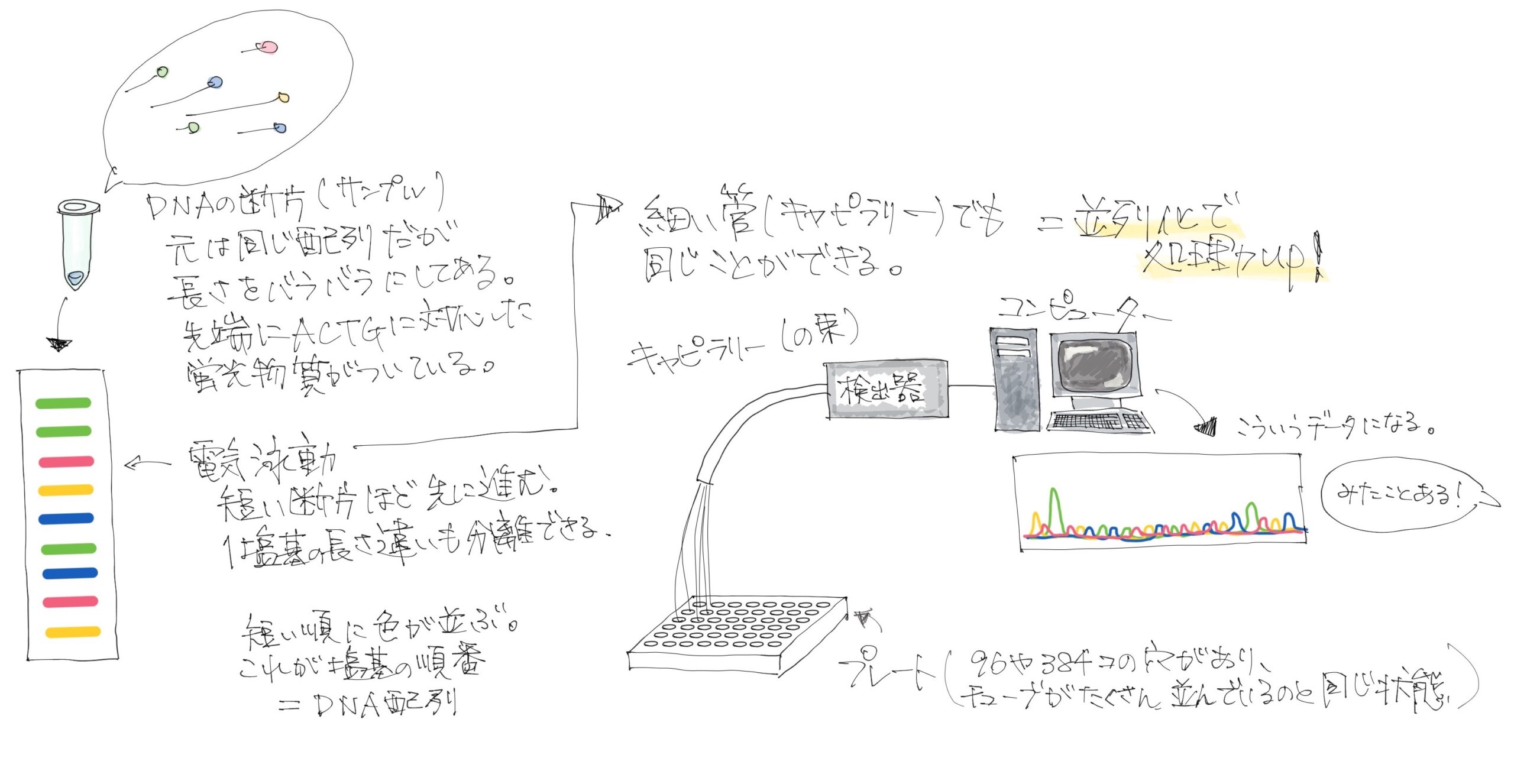
まぁ、そう言われればそうですね。
今、主力で使われるNGSは、もう発想が違う。キャピラリーとかなくて平面なスライドです。スライド上に足場になる短いDNAの断片が貼り付けられていて、サンプルの中のDNAがその断片にくっついて、それを読んでいくんです。
すごい数になりそう。
最新機種だと100億サンプルとかですね。桁とか単位が違うレベルではありません。キャピラリーの場合は、384サンプルくらいが現実的にハンドルできるサイズでしたから、ものすごい飛躍です。
たしか写真を撮ってるんですよね。
そうです。このスライド上にサンプルのDNAを固定して、対となる蛍光標識されたDNAを1塩基ずつくっつけていってその都度写真を撮ります。スライド全体の写真を撮るので、100億サンプルの超並列化解析です。これを何サイクルもやると、最初はA、次はT…という感じにデータが100億サンプル同時に得られます。
画像解析ができるようになったからこそ、できることですね。コンピューター様様。
これがいわゆるNGS=次世代DNAシーケンサー。多く使われているのはイルミナという会社の装置ですね。

もともとはソレクサという会社の技術だったんですよね。個人的にはソレクサの名前が忘れ去られているのは残念ですけどね。
ここまではDNAをかなり短い断片にして読む方式で、先ほど言った、似たような配列の問題は解決しなかったんですが、Pacific Biosciences社やOxford Nanopore社からさらに新しい技術が出てきて、これは長いDNA配列が読めるんです。第3世代とか第4世代とか言われています。
ずーっと次世代…。

Nanoporeは手のひらに乗るサイズです。これだとどこにでも持って行けるので極地でもシーケンスができる。国際宇宙ステーションでゲノムを読んだりしてましたね。
それにしても、小さいなぁ…。
例えばブラジルのジャングルの奥地に行って面白い生き物を取ってきても、サンプルの保存や検疫、野生動植物保護の観点から、日本には持ち込めないんです。そういう極地での使用、フィールドサイエンスにはけっこうニーズが高い技術でもあるんです。
新型コロナのPCRもワクチンも
よく考えたら、今はゲノムの情報があるとかゲノムが読めるとかというのが前提になってることってたくさんありますね。
そうです。たとえば、新型コロナのゲノムがなぜ1日で解読できるかというと、次世代DNAシーケンサーがあるからなわけです。
PCRも、そもそも配列わからないと設計できないですもんねぇ。
変異株という話もありますけど、これもゲノムの全配列を読んでるからわかるわけです。
でも、ヒトゲノムのように、ドラフトってわけにもいかないですよね。
ウイルスゲノムはかなり小さいので、ヒトほど大変ではないですね。ただ、ウイルスが扱える施設でないといけないですけど。
他の生物もまだゲノムが読めていない生物もいますよね。
新しく読もうとするとヒトゲノムのときと同じハードルがやっぱりあります。この配列、どこのだ?配列が全然つながらない!みたいな。なので、読んだゲノム配列がどのくらい確からしいのかを簡単に評価出来るツールを作ってWebで公開しています。
え?初めて読む場合だと、正解ってわからないですよね?
進化的な観点から評価するんです。進化的に我々ヒトは、哺乳類だし、脊椎動物ですよね。チンパンジーとの共通祖先もいるし、魚との共通祖先もいるし、という、進化の枝分かれがあってきているわけです。そうすると、本質的には共通祖先が持っていた遺伝子というのは、現代の全ての脊椎動物、それはもうカエルだろうが、魚だろうが、ヒトだろうが、ネズミだろうが、持っているはず、ということになります。進化の過程で、哺乳類ではこの遺伝子は失われたんだなということとか、逆に重複して新たな遺伝子が出来たんだなということも、複数の種のゲノムを解読するとわかってきます。そういったデータベース、例えば「哺乳類なら必ず持っているはずの遺伝子セット」が出来れば、全く新規の哺乳類ゲノムであっても、ある程度の評価ができます。このアイデアや手法は以前から報告されているのですが、解析が難しいので、Webツールにしてみました。
なるほど。いろんな種の配列を読む、というのはそういう生物学的な面白さもあるんですね。そして、そんなウェブツールを公開してるんですね。知らなかった。
このラボは、センターのコアファシリティとしての役割もあります。お話ししたように、今の生物学にとってゲノムや遺伝子の解析というのは不可欠になってきています。一方で、技術の進展が速く、幅も広がってきているので、それぞれのラボや研究員でそれらの情報を把握して、技術を習得して実際に解析するというのはかなり難しくなってきています。
確かに、技術を追いかけるだけでも大変そうですね。
しかも、それを解析するというコンピューターサイドの知識、技術も必要になります。一見、クオリティが低いデータであっても、よくよく解析し直してみると、必要な情報の部分は十分なクオリティがあって、データ解析には支障ない、なんてこともあります。
そういう支援もしてるんですか?
かなり丁寧に対応していますよ。うちは「愛のある」コアファシリティなんです(笑)

編集後記
なんだか、シーケンサーの技術やゲノム解析の歴史を一気に見た気分。ゲノムの情報がないと、新型コロナのPCRもワクチンもできないというのを改めて認識すると、こういう基礎技術が実際に世の中の役に立っているというのを実感しますね。